
Decoding Nature’s Chemistry
Fungi: A new starting point for drug discovery
Dear SoTA,
The future of drug discovery lies in learning from natural evolution, shaped by billions of years of optimization, not in forcing synthetic chemistry on complex biology. Nature offers immense medicinal potential, yet 99% of its chemistry remains unexplored. Unlocking this frontier starts with the ability to rapidly analyze natural molecules at scale.
Expanding chemical space for drug discovery
Small molecule therapeutics remain the cornerstone of modern medicine. Yet most new drugs are built from a limited set of known chemical scaffolds that are easy to synthesize. This has kept discovery efforts clustered in a narrow region of chemical space, repeatedly optimizing known, low-complexity structures rather than exploring novel ones.
Meanwhile, nature operates in a far broader chemical universe. Over billions of years, it has generated complex, functional molecules far beyond the diversity of what humans can make. About half of all approved small-molecule drugs are in fact structurally inspired by natural designs, with evolution optimizing these molecules for biological function.
Many multi-billion-dollar drugs trace their origins to natural molecules: statins (~$17B) for lowering cholesterol, Penicillin (~$10B) for antibiotics, Cyclosporine A (~$3B) for immunosuppression, Fingolimod (~$2B) for multiple sclerosis, and Paclitaxel (~$6B) for cancer therapy. Natural molecules also have faster developmental timelines and a 30% higher chance of passing clinical trials.
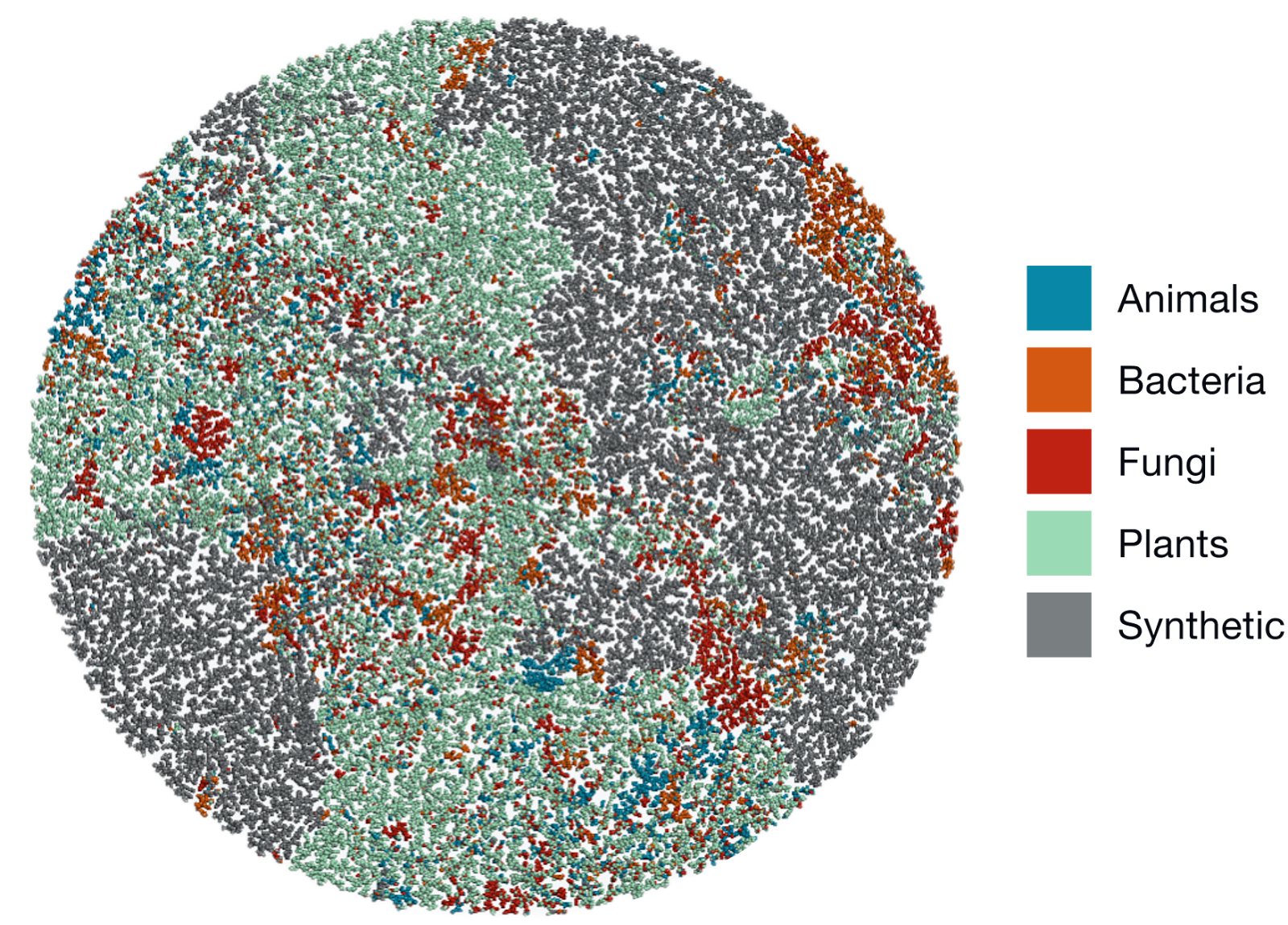
The visualization below shows how natural molecules (from animals, bacteria, fungi, and plants) occupy distinct regions of chemical space compared to synthetic ones. Each dot represents a molecule, clustered by similarity.
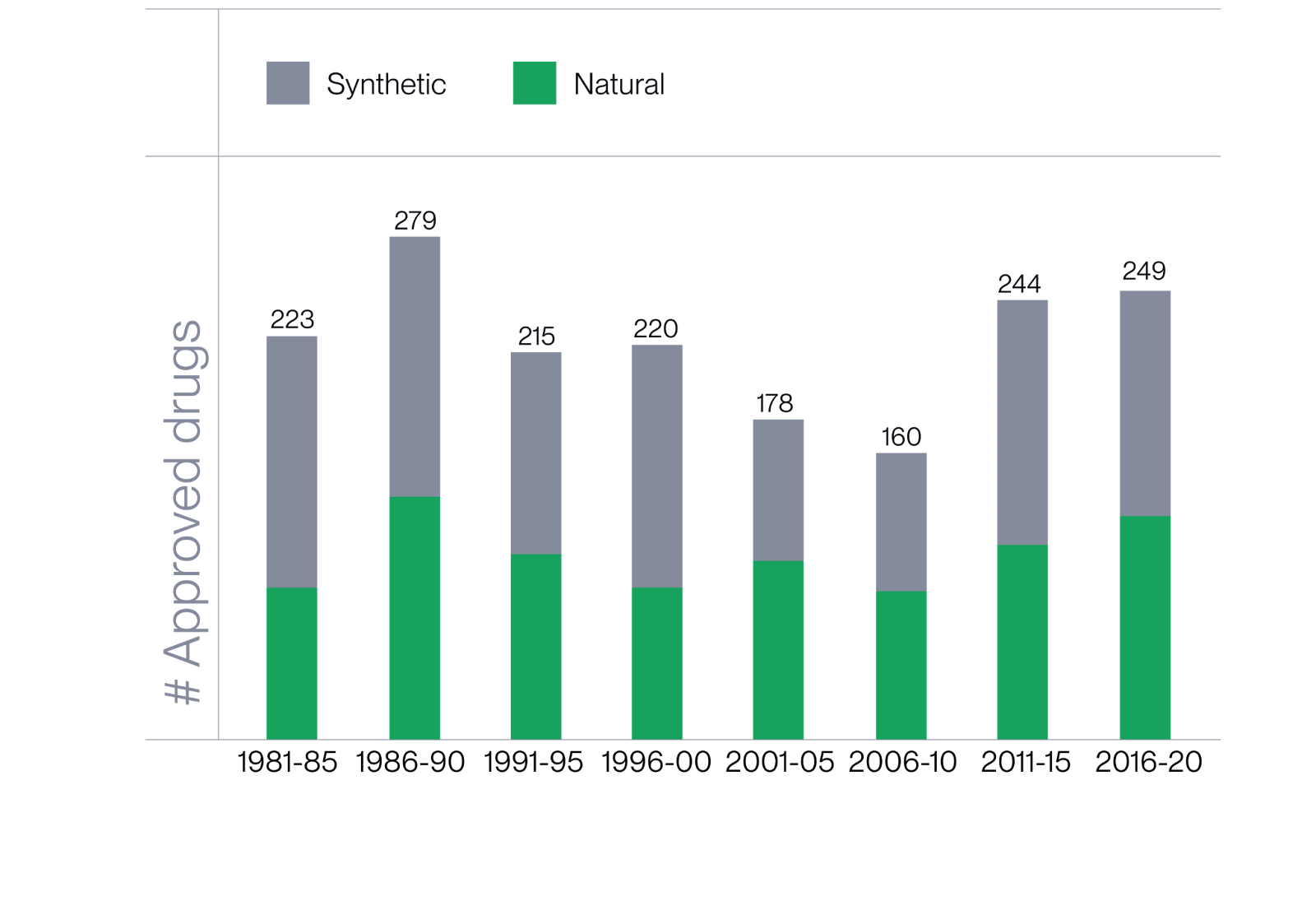
While screening nature for new molecules has sharply declined since the 1980s due to slow, manual discovery workflows and limited structural identification tools, advances in machine learning and analytical chemistry now make it possible to decode nature’s chemistry at scale. This unlocks vast, bioactive regions of chemical space that were previously out of reach for drug discovery.
Gaia-01: AI for molecular structure prediction from spectra
The key step for knowing what molecules are in a natural sample, and whether they are interesting for drug discovery, is decoding their chemical structure. Mass spectrometry is the fastest and most sensitive method for profiling molecules from natural samples, and its resolution is now orders of magnitude higher than during past large-scale screening efforts. The technology works by breaking molecules into fragments and measuring their mass-to-charge ratios, the patterns of which can be used to infer a molecule’s chemical structure. Recent advances now allow fine-grained distinction between closely related molecules and reconstruction of complex molecular structures directly from natural samples. Modern machine learning transforms this detection tool into a predictive tool for inferring molecular structures directly from complex mixtures of natural molecules. This task would otherwise require lengthy isolation and purification of each molecule, followed by structure determination using more advanced tools such as nuclear magnetic resonance (NMR).
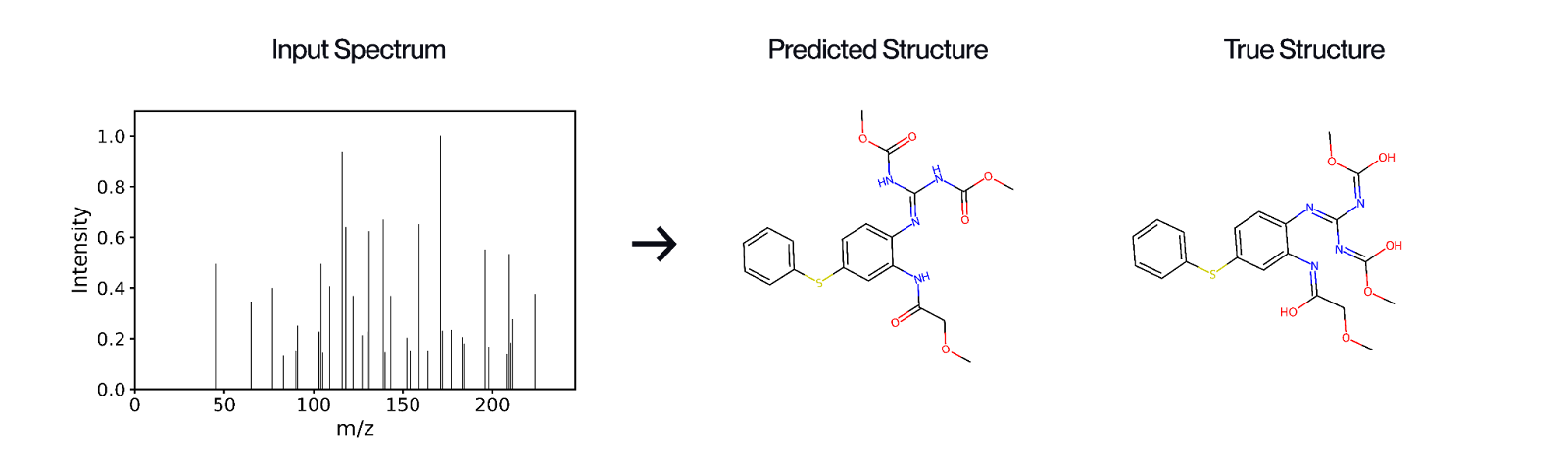
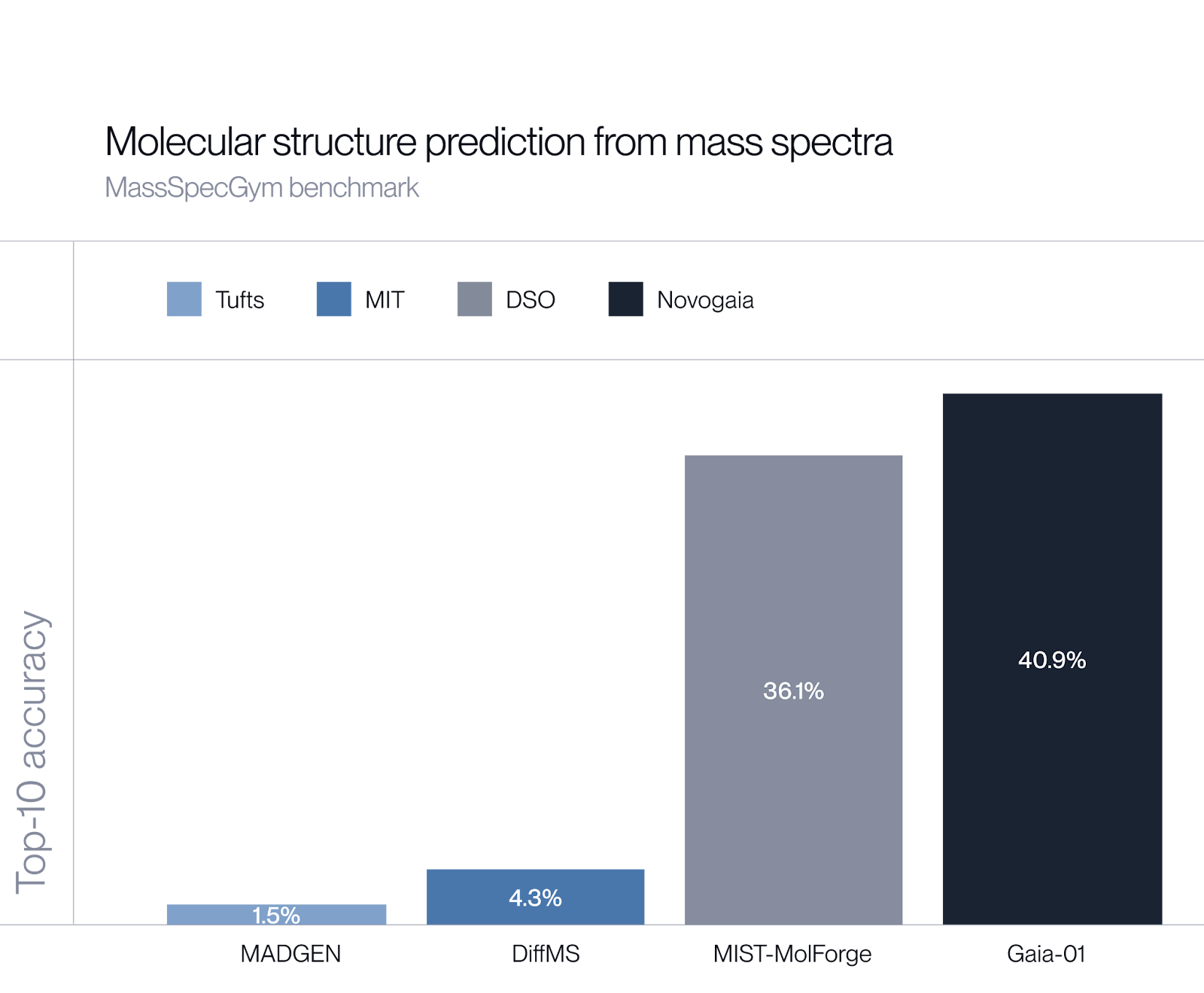
We’ve built Gaia-01, a 1-billion-parameter autoregressive transformer model that predicts molecular structures directly from mass spectrometry data and achieves a 13% performance improvement over the current state of the art. More in our blogpost.
Gaia-01 advances two critical capabilities:
New chemical starting points for drug discovery, directly from nature
Gaia-01 allows us to rapidly identify molecules with drug-like properties from natural samples. From these natural molecules, we can design synthetic analogues for testing against therapeutic targets, bridging nature’s molecular diversity with modern medicinal chemistry.A vastly expanded data foundation for generative molecule design
Current generative small molecule models repurpose known compounds due to limited data. Gaia-01 can recover molecular structures hidden in millions of publicly available mass spectral datapoints, expanding the set of known natural molecules by up to 100-fold. This opens the door to generative models that learn not just from human-made chemistry, but from nature’s own design principles.
At Novogaia, we apply these technologies to decode fungal chemistry. Fungi remain one of nature’s richest but most unexploited sources of pharmacologically active molecules. Our mission is to unlock a new era in drug discovery from fungi by using AI to systematically uncover their molecular diversity and translate it into new therapeutic breakthroughs. To make that happen, we’re building a broader AI-driven discovery pipeline that brings this technology fully to life.
Nature has far more to offer than we’ve ever been able to see. With the right tools, we can finally start to look.
Yours,
Fascinating work on expanding the compond library for early discovery. That 30% trial sucess rate advantage for natural mollecules is understated as a risk reducer in trial design. I'd be curious how the bioavailability profiles of your fungal-derived candidates compare to synthetics when factoring in absorption and metabolic stability for Phase I planning.